EFFICACY—DOR
VITRAKVI®: REMARKABLE RESPONSES DEMONSTRATED TO LAST1
INITIAL DATA SET (N=55)a
3 OUT OF 4 PATIENTS HAD AN OBJECTIVE RESPONSE TO VITRAKVI1
Overall Response Rate1,2

 overall response rate")
Overall Response Rate1,2
 overall response rate")
bCI, confidence interval; CR, complete response; ORR, overall response rate; PR, partial response.
c5% were pathological complete response.1,3 Patients undergoing a surgical resection whose postoperative pathologic assessment showed no viable tumor cells and negative margins were pathological complete responders provided that no other sites of disease were present.1
aJuly 2019 cutoff.1
bCI, confidence interval; CR, complete response; ORR, overall response rate; PR, partial response.
c5% were pathological complete response.1,3 Patients undergoing a surgical resection whose postoperative pathologic assessment showed no viable tumor cells and negative margins were pathological complete responders provided that no other sites of disease were present.1
INITIAL DATA SET (N=55)a
THE TRK* INHIBITOR THAT DELIVERS A MEDIAN DORb LASTING NEARLY 3 YEARS1
Duration of Response (n=41/55)1,2,c

Duration of Response (n=41/55)1,2,c
bCI, confidence interval; DOR, duration of response; NE, not evaluable.
cKaplan-Meier estimate.1
Results in an expanded data setd (N=244): median DOR of 38.4 months.2
VITRAKVI was studied across multiple lines of therapy1,2
- 20% of patients received no prior systemic therapy2
- 45% received 1 or 2 prior systemic therapies2
- 35% received ≥3 prior systemic therapies1,2
- 63% of patients with a response had an observed DOR >12 months1
- 49% of patients with a response had an observed DOR >24 months1
In an expanded data set (N=244) not reviewed by the FDA, 18 patients were evaluable with brain metastases at baseline2,e:

July 2022 cutoff.
mDORb: 14.8 MONTHS2:
(95% CI: 59%, 96%) (n=15/18)
- 60% of patients with a response had an observed DOR ≥6 months
- Median follow-up of 23 months (25th, 75th percentiles: 8.2, 23.3)2
VITRAKVI is approved under accelerated approval based on overall response rate and duration of response. Continued approval for VITRAKVI may be contingent upon verification and description of clinical benefit in confirmatory trials.1
STUDY DESIGN:
Pooled efficacy analysis was based on 3 multicenter, open-label, single-arm clinical studies in adult and pediatric patients with unresectable or metastatic solid tumors with an NTRKb gene fusion. All patients were required to have progressed following systemic therapy for their disease if available or would have required surgery with significant morbidity for locally advanced disease. Major efficacy outcome measures were ORRb and DOR as determined by BIRCb according to RECISTb v1.1.1
+ denotes ongoing response.
*TRK, tropomyosin receptor kinase.
aJuly 2019 cutoff.1
bBIRC, blinded independent review committee; CI, confidence interval; DOR, duration of response; FDA, US Food and Drug Administration; IRC, independent review committee; mDOR, median duration of response; NE, not evaluable; NTRK , neurotrophic receptor tyrosine kinase; ORR, overall response rate; RECIST, Response Evaluation Criteria in Solid Tumors.
cKaplan-Meier estimate.1
dBased on an IRCa; July 2021 cutoff.3
eBased on an IRC. Data set included the initial 55 patients plus an additional 189 patients; July 2022 cutoff.1,2
FE | Patient Cases
PATIENT CASES
Lung
76-YEAR-OLD
FEMALE
WITH
BRAIN METASTASES
30-YEAR-OLD
FEMALE
WITH BONE
METASTASES
Thyroid
33-YEAR-OLD
MALE
56-YEAR-OLD
FEMALE
WITH
MULTIPLE
METASTASES
Pediatric
5-MONTH-OLD WITH
INFANTILE
FIBROSARCOMA
PATIENT CASE:
INFANTILE FIBROSARCOMA
OF SOFT TISSUE1
- 5-month-old with IFSa
- Progression after chemotherapy
- Confirmed PRa after 4 cycles of VITRAKVI® (larotrectinib)
- Referred for definitive limb-sparing surgery after 6 cycles
of VITRAKVI; achieved pCRa,b
This is a response from 1 patient. Different patients may have different responses to therapy.
This patient profile is adapted from a real patient case.
Clinical presentation1
- Diagnosed with localized IFS of the soft tissue of the forearm
Prior treatments and outcomes1
- Patient progressed after 2 cycles of vincristine and actinomycin D
- He was subsequently placed on vincristine, actinomycin D, and cyclophosphamide; however, his response remained inadequate to allow for limb-sparing surgery
aIFS, infantile fibrosarcoma; pCR, pathological complete response; PR, partial response.
bPatients undergoing a surgical resection whose postoperative pathologic assessment showed no viable tumor cells and negative margins were pathological complete responders provided that no other sites of disease were present.2
Advise women not to breastfeed during treatment with VITRAKVI and for 1 week after the final dose.2
Testing1
- ETV6a-NTRK3a fusion identified with DNAa NGSa panel testing
aDNA, deoxyribonucleic acid; ETV6, ETS variant 6; IFS, infantile fibrosarcoma; NTRK, neurotrophic receptor tyrosine kinase; NGS, next-generation sequencing; pCR, pathological complete response; PR, partial response.
bPatients undergoing a surgical resection whose postoperative pathologic assessment showed no viable tumor cells and negative margins were pathological complete responders provided that no other sites of disease were present.2
VITRAKVI treatment1
- VITRAKVI 100 mg twice daily orally
Response to VITRAKVI1
- Patient achieved a confirmed partial response after 4 cycles of VITRAKVI with a 45% reduction in tumor burden
- Following 6 cycles of VITRAKVI, the patient was referred for definitive limb-sparing surgery
- Pathology revealed a complete pathologic response and clear resection margins with scar tissue noted
aIFS, infantile fibrosarcoma; pCR, pathological complete response; PR, partial response.
bPatients undergoing a surgical resection whose postoperative pathologic assessment showed no viable tumor cells and negative margins were pathological complete responders provided that no other sites of disease were present.2
Reduction in tumor burden1

MRIa imagery of the brain. Red line indicates the maximum dimension.1

Reduction in tumor burden1

MRIa imagery of the brain. Red line indicates the maximum dimension.1
aMRI, magnetic resonance imaging.

Pre- and post-treatment imaging, by DuBois SG et al, is licensed under CC BY 4.0.
aIFS, infantile fibrosarcoma; MRI, magnetic resonance imaging; pCR, pathological complete response; PR, partial response.
bPatients undergoing a surgical resection whose postoperative pathologic assessment showed no viable tumor cells and negative margins were pathological complete responders provided that no other sites of disease were present.2
References
- DuBois SG, Laetsch TW, Federman N, et al. The use of neoadjuvant larotrectinib in the management of children with locally advanced TRK fusion sarcomas. Cancer. 2018;124(21):4241-4247. Return to content
- VITRAKVI [package insert]. Whippany, NJ: Bayer HealthCare Pharmaceuticals, Inc.; November 2023. Return to content
aIFS, infantile fibrosarcoma; pCR, pathological complete response; PR, partial response
bPatients undergoing a surgical resection whose postoperative pathologic assessment showed no viable tumor cells and negative margins were pathological complete responders provided that no other sites of disease were present.2
PATIENT CASE:
METASTATIC, RAIa-REFRACTORY,
PAPILLARY THYROID CANCER1
- 56-year-old female with PTCa that had metastasized to multiple sites, including the brain
- Progression after first- and second-line systemic treatment
- PRa after 4 weeks on VITRAKVI® (larotrectinib); CRa after 8 weeks
- At last assessment, complete response was sustained over an 11-month period
This is a response from 1 patient. Different patients may have different responses to therapy.
This patient profile is adapted from a real patient case.
Clinical presentation1
- Patient was assessed to have metastatic, rapidly progressive, RAl-refractory PTC based on imaging that revealed multiple metastatic sites, including the neck, mediastinum, lung, scalp, and bones
- Biopsy of a scalp lesion helped to confirm the PTC metastasis
- Metastases to the brain and liver were observed after progression on systemic treatments
Prior treatments and outcomes1
- Prior treatments included surgery, RAI, a combination of zoledronic acid and sorafenib, external beam radiotherapy, and then lenvatinib
aCR, complete response; PR, partial response; PTC, papillary thyroid cancer; RAI, radioactive iodine.
Advise women not to breastfeed during treatment with VITRAKVI and for 1 week after the final dose.2
Testing1
- With no further treatment options, the decision was made to perform genomic testing. NGSa testing was performed on a scalp biopsy and revealed an ETV6a-NTRK3a gene fusion. The scalp biopsy had been performed 1 year earlier.
NCCNa Clinical Practice Guidelines in Oncology (NCCN Guidelines®) recommend genomic testing to identify actionable mutations, including NTRK, for advanced, progressive, or threatening thyroid carcinoma.3
aCR, complete response; ETV6, ETS variant 6; NCCN, National Comprehensive Cancer Network® (NCCN®); NGS, next-generation sequencing; NTRK, neurotrophic receptor tyrosine kinase; PR, partial response; PTC, papillary thyroid cancer; RAI, radioactive iodine.
VITRAKVI treatment1
- VITRAKVI 100 mg twice daily orally
Response to VITRAKVI1
- After 8 weeks on VITRAKVI, all target lesions had disappeared, demonstrating a complete response
- The complete response was sustained over 11 months of treatment with VITRAKVI
- Patient experienced Grade 1 fatigue and mild hepatic enzyme elevation
aCR, complete response; PR, partial response; PTC, papillary thyroid cancer; RAI, radioactive iodine.
Response in primary and metastatic lesions1

MRIa imagery of the lungs. Arrow indicates large target lesion in left lung.1
Response in primary and metastatic lesions1
SCAN 1: LUNG

MRIa imagery of the brain. Arrow indicates large target lesion in left lung.1
aMRI, magnetic resonance imaging.









Images courtesy of Dr Fabian Pitoia.
aCR, complete response; MRI, magnetic resonance imaging; PR, partial response; PTC, papillary thyroid cancer; RAI, radioactive iodine.
References
- Pitoia F. Complete response to larotrectinib treatment in a patient with papillary thyroid cancer harboring an ETV6-NTRK3 gene fusion. Clin Case Rep. 2021;9(4):1905-1912. Return to content
- VITRAKVI [package insert]. Whippany, NJ: Bayer HealthCare Pharmaceuticals, Inc.; November 2023. Return to content
- Referenced with permission from the NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for Thyroid Carcinoma V.3.2024. © National Comprehensive Cancer Network, Inc. 2024. All rights reserved. Accessed July 26, 2024. To view the most recent and complete version of the guidelines, go online to NCCN.org. NCCN makes no warranties of any kind whatsoever regarding their content, use or application and disclaims any responsibility for their application or use in any way. Return to content
aCR, complete response; PR, partial response; PTC, papillary thyroid cancer; RAI, radioactive iodine.
Patient Case - 76 year old - landing
PATIENT CASE:
NSCLCa PATIENT WITH BRAIN
METASTASES1-3
- 76-year-old female with NSCLC that metastasized to the brain and liver
- No prior systemic treatment before trial enrollment
- PRa of primary lung nodules to VITRAKVI® (larotrectinib) confirmed at 2 months1
This is a response from 1 patient. Different patients may have different responses to therapy.
This patient profile is adapted from a real patient case.
Clinical presentation1,2
- Presented with a persistent cough, fatigue, and anorexia
- Imaging identified bilateral lung nodules with metastases to the brain
- Endobronchial biopsy confirmed lung adenocarcinoma
Prior treatments and outcomes3
- No prior surgery or radiation
- Patient elected not to undergo platinum doublet chemotherapy treatment for her lung cancer in front line
aNSCLC, non-small cell lung cancer; PR, partial response.
Advise women not to breastfeed during treatment with VITRAKVI and for 1 week after the final dose.4
Testing1
- Initial DNAa-based NGSa testing was negative for NTRKa gene fusions
- Subsequent RNAa-based NGS testing identified EPS15a-NTRK1 gene fusion
NCCNa Clinical Practice Guidelines in Oncology (NCCN Guidelines®) recommend molecular testing that includes NTRK gene fusion for advanced or metastatic NSCLC.5
aDNA, deoxyribonucleic acid; EPS15, epidermal growth factor receptor pathway substrate; NCCN, National Comprehensive Cancer Network® (NCCN®); NGS, next-generation sequencing; NSCLC, non-small cell lung cancer; NTRK, neurotrophic receptor tyrosine kinase; PR, partial response; RNA, ribonucleic acid.
VITRAKVI treatment2
- VITRAKVI 100 mg twice daily orally
Response to VITRAKVI1,2
- Partial response to treatment that was confirmed at 2 months (34% tumor reduction)
- Grade 1 cough and Grade 2 fatigue
- Patient remains on VITRAKVI after more than 4 months of treatment
aNSCLC, non-small cell lung cancer; PR, partial response.
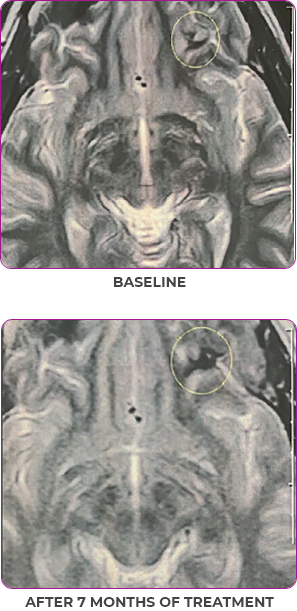
Response in primary and metastatic lesions1



MRIa imagery of the brain. Green circles indicate baseline brain metastases.1
Response in primary and metastatic lesions1
SCAN 2: BRAIN

MRIa imagery of the brain. Green circles indicate baseline brain metastases.1
aMRI, magnetic resonance imaging.

MRIa imagery of the brain. Green voxels indicate decreased burden of metastatic disease; further quantified by accompanying measurement in green.1
Response in primary and metastatic lesions1
SCAN 3: BRAIN

MRIa imagery of the brain. Green voxels indicate decreased burden of metastatic disease; further quantified by accompanying measurement in green.1
aMRI, magnetic resonance imaging.



Pre- and post-treatment imaging, by Rosen EY et al, is licensed under Creative Commons License CC BY 4.0.
aNSCLC, non-small cell lung cancer; PR, partial response.
References
- Rosen EY, Schram AM, Young RJ, et al. Larotrectinib demonstrates CNS efficacy in TRK fusion-positive solid tumors. JCO Precis Oncol. 2019;3:1-5. Return to content
- Lassen U, Albert CM, Kummar S, et al. Larotrectinib efficacy and safety in TRK fusion cancer: an expanded clinical dataset showing consistency in an age and tumor agnostic approach. Presented at: European Society for Medical Oncology Congress; October 19-23, 2018; Munich, Germany. Return to content
- Drilon A, Moreno V, Patel J, et al. Efficacy and safety of larotrectinib in patients with tropomyosin receptor kinase (TRK) fusion lung cancer. Poster presented at: ESMO Virtual Congress 2020; September 19-21, 2020. Return to content
- VITRAKVI [package insert]. Whippany, NJ: Bayer HealthCare Pharmaceuticals, Inc.; November 2023. Return to content
- Referenced with permission from the NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for Non-Small Cell Lung Cancer V.7.2024. © National Comprehensive Cancer Network, Inc. 2024. All rights reserved. Accessed July 26, 2024. To view the most recent and complete version of the guidelines, go online to NCCN.org. NCCN makes no warranties of any kind whatsoever regarding their content, use or application and disclaims any responsibility for their application or use in any way. Return to content
aNSCLC, non-small cell lung cancer; PR, partial response.
PATIENT CASE:
NSCLCa PATIENT WITH BRAIN
METASTASES1-3
- 76-year-old female with NSCLC that metastasized to the the brain and liver
- No prior systemic treatment before trial enrollment
- PRa of primary lung nodules to VITRAKVI® (larotrectinib) confirmed at 2 months1
This is a response from 1 patient. Different patients may have different responses to therapy.
This patient profile is adapted from a real patient case.
Clinical presentation1,2
- Presented with a persistent cough, fatigue, and anorexia
- Imaging identified bilateral lung nodules with metastases to the brain
- Endobronchial biopsy confirmed lung adenocarcinoma
Prior treatments and outcomes3
- No prior surgery or radiation
- Patient elected not to undergo platinum doublet chemotherapy treatment for her lung cancer in front line
aNSCLC, non-small cell lung cancer; PR, partial response.
Advise women not to breastfeed during treatment with VITRAKVI and for 1 week after the final dose.4
Testing1
- Initial DNAa-based NGSa testing was negative for NTRKa gene fusions
- Subsequent RNAa-based NGS testing identified EPS15a-NTRK1 gene fusion
NCCNa Clinical Practice Guidelines in Oncology (NCCN Guidelines®) recommend molecular testing that includes NTRK gene fusion for advanced or metastatic NSCLC.5
aDNA, deoxyribonucleic acid; EPS15, epidermal growth factor receptor pathway substrate; NCCN, National Comprehensive Cancer Network® (NCCN®); NGS, next-generation sequencing; NSCLC, non-small cell lung cancer; NTRK, neurotrophic receptor tyrosine kinase; PR, partial response; RNA, ribonucleic acid.
VITRAKVI treatment2
- VITRAKVI 100 mg BIDa orally
Response to VITRAKVI1,2
- Partial response to treatment that was confirmed at 2 months (34% tumor reduction)
- Grade 1 cough and Grade 2 fatigue
- Patient remains on VITRAKVI after more than 4 months of treatment
aBID, twice daily; NSCLC, non-small cell lung cancer; PR, partial response.
Response in primary and metastatic lesions1
MRIa imagery of the brain. Green circles indicate baseline brain metastases.1

Response in primary and metastatic lesions1
SCAN 2: BRAIN
MRIa imagery of the brain. Green circles indicate baseline brain metastases.1
aMRI, magnetic resonance imaging.
MRIa imagery of the brain. Green voxels indicate decreased burden of metastatic disease; further quantified by accompanying measurement in green.1
Response in primary and metastatic lesions1
SCAN 3: BRAIN
MRIa imagery of the brain. Green voxels indicate decreased burden of metastatic disease; further quantified by accompanying measurement in green.1
aMRI, magnetic resonance imaging.

Pre- and post-treatment imaging, by Rosen EY et al, is licensed under Creative Commons License CC BY 4.0.
aMRI, magnetic resonance imaging; NSCLC, non-small cell lung cancer; PR, partial response.
References
- Rosen EY, Schram AM, Young RJ, et al. Larotrectinib demonstrates CNS efficacy in TRK fusion-positive solid tumors. JCO Precis Oncol. 2019;3:1-5. Return to content
- Lassen U, Albert CM, Kummar S, et al. Larotrectinib efficacy and safety in TRK fusion cancer: an expanded clinical dataset showing consistency in an age and tumor agnostic approach. Presented at: European Society for Medical Oncology Congress; October 19-23, 2018; Munich, Germany. Return to content
- Drilon A, Moreno V, Patel J, et al. Efficacy and safety of larotrectinib in patients with tropomyosin receptor kinase (TRK) fusion lung cancer. Poster presented at: ESMO Virtual Congress 2020; September 19-21, 2020. Return to content
- VITRAKVI [package insert]. Whippany, NJ: Bayer HealthCare Pharmaceuticals, Inc.; December 2022. Return to content
- Referenced with permission from the NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for Non-Small Cell Lung Cancer V1.2023. © National Comprehensive Cancer Network, Inc. 2023. All rights reserved. Accessed January 4, 2023. To view the most recent and complete version of the guidelines, go online to NCCN.org. NCCN makes no warranties of any kind whatsoever regarding their content, use or application and disclaims any responsibility for their application or use in any way. Return to content
aNSCLC, non-small cell lung cancer; PR, partial response.
PATIENT CASE:
METASTATIC NSCLC1,a
- 30-year-old female
- Progression on first-line doublet chemotherapy
- PRa after 6 weeks of treatment with VITRAKVI® (larotrectinib)
- Complete clinical response achieved by 12 months with residual scarring
This is a response from 1 patient. Different patients may have different responses to therapy.
This patient profile is adapted from a real patient case.
Clinical presentation1
- Nonsmoker and a history of atopic eczema
- Presented with dyspnea and pleural effusion
- Bone-scan imaging revealed 3 metastatic lesions of the pelvis, in the sacrum, acetabulum, and ischial bones, which were asymptomatic
- Diagnosed with metastatic adenocarcinoma of the right lung in March 2017
Prior treatments and outcomes1
- 4 cycles of cisplatin plus pemetrexed, followed by pemetrexed maintenance for 1 year. Partial remission was achieved and treatment was discontinued at patient request
- Progression recurred 5 months later
- lmmunotherapy was considered, but not selected due to patient’s concern about autoimmune AEsa from preexisting atopic eczema
aAE, adverse event; NSCLC, non-small cell lung cancer; PR, partial response.
Advise women not to breastfeed during treatment with VITRAKVI and for 1 week after the final dose.2
Testing1
- NGSa testing was not routine at the time of diagnosis. Instead, molecular testing for EGFRa mutations (real-time PCRa), ALK,a and ROS1a (IHCa) was performed and showed no targetable abnormalities. PD-L1a expression was detected in approximately 1% of tumor cellsb
- NTRKa testing was not performed until 3 months after disease progression. Patient was found to be NTRK gene fusion–positive based on both IHC and NGS. TPM3a-NTRK1 gene fusion was identified as the oncogenic driver
NCCNa Clinical Practice Guidelines in Oncology (NCCN Guidelines®) recommend molecular testing that includes NTRK gene fusion for advanced or metastatic NSCLC.3
aALK, anaplastic lymphoma kinase; EGFR, epidermal growth factor receptor; FDA, US Food and Drug Administration; IHC, immunohistochemistry; NCCN, National Comprehensive Cancer Network® (NCCN®); NGS, next-generation sequencing; NSCLC, non-small cell lung cancer; NTRK, neurotrophic receptor tyrosine kinase; PCR, polymerase chain reaction; PD-L1, programmed death-ligand 1; PR, partial response; ROS1, c-ros oncogene 1; TPM3, tropomyosin 3.
bSelect patients for therapy based on FDAa-approved test.1
VITRAKVI treatment1
- VITRAKVI 100 mg twice daily orally
Response to VITRAKVI1
- Partial response and symptom improvement confirmed by chest X-rays after <1 month of treatment
- Imaging performed 6 weeks into treatment revealed considerable decrease in the size of both target lesions
- No treatment-related AEsa were reported
- Complete clinical response achieved by 12 months with residual scarring
- Patient was symptom free and remained on VITRAKVI
aAE, adverse event; NSCLC, non-small cell lung cancer; PR, partial response.
Response in primary and metastatic lesions1
Lung imaging of primary tumors.

Response in primary and metastatic lesions1
Lung imaging of primary tumors.
SCAN 1: LUNG


Response in primary and metastatic lesions1
Lung imaging of primary tumors.
SCAN 2: LUNG AND BONE



Images courtesy of Dr Maximilian Hochmair.
aNSCLC, non-small cell lung cancer; PR, partial response.
References
- Blay J-Y, Brose MS, García-Foncillas J, Hochmair MJ, Vainer G. Precision medicine in real-world clinical practice: perspectives from a multidisciplinary team. Virtual presentation at: European Society for Medical Oncology Virtual Congress 2020 Industry Satellite Symposium. On demand from September 14, 2020. Return to content
- VITRAKVI [package insert]. Whippany, NJ: Bayer HealthCare Pharmaceuticals, Inc.; November 2023. Return to content
- Referenced with permission from the NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for Non-Small Cell Lung Cancer V.7.2024. © National Comprehensive Cancer Network, Inc. 2024. All rights reserved. Accessed July 26, 2024. To view the most recent and complete version of the guidelines, go online to NCCN.org. NCCN makes no warranties of any kind whatsoever regarding their content, use or application and disclaims any responsibility for their application or use in any way. Return to content
aNSCLC, non-small cell lung cancer; PR partial response.
PATIENT CASE:
METASTATIC, RAIa-REFRACTORY,
PAPILLARY THYROID CANCER1,2
- 33-year-old male
- Progression on RAI and first line systemic therapy
- PRa after 2 cycles of VITRAKVI® (larotrectinib)
- Confirmed partial response on VITRAKVI lasting 55 cycles
This is a response from 1 patient. Different patients may have different responses to therapy.
This patient profile is adapted from a real patient case.
Clinical presentation1
- Initially diagnosed at age 27
- Bulky disease in neck and thoracic involvement; metastatic, RAI-refractory papillary thyroid cancer
Prior treatments and outcomes1
- 5 prior surgeries
- 2 prior RAI treatments
- Systemic therapy with pazopanib and trametinib
aPR, partial response; RAI, radioactive iodine.
Testing1,2
- ETV6a-NTRK3a gene fusion was detected during disease progression on systemic therapy via NGS
NCCNa Clinical Practice Guidelines in Oncology (NCCN Guidelines®) recommend genomic testing to identify actionable mutations, including NTRK, for advanced, progressive, or threatening thyroid carcinoma.3
aETV6, ETS variant 6; NCCN, National Comprehensive Cancer Network® (NCCN®); NTRK, neurotrophic receptor tyrosine kinase; PR, partial response; RAI, radioactive iodine.
VITRAKVI treatment1
- VITRAKVI 100 mg twice daily orally
Response to VITRAKVI1,2
- Confirmed partial response (92.6% reduction in target lesions)
- Treatment continued for 55 cycles
- VITRAKVI was well tolerated; patient did not have to discontinue due to AEa
- No treatment interruptions or dose reductions
aAE, adverse event; PR, partial response; RAI, radioactive iodine.
Response in primary and metastatic lesions1




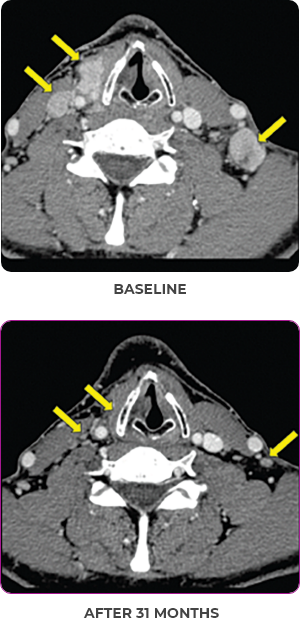

Images courtesy of Dr Steven Waguespack.
aPR, partial response; RAI, radioactive iodine.
References
- Waguespack SG, Drilon A, Farago AF, et al. Treatment of advanced TRK fusion thyroid cancer with larotrectinib. Presented at: 42nd Annual Meeting of the European Thyroid Association; September 7-10, 2019; Budapest, Hungary. Return to content
- Data on file. Bayer HealthCare Pharmaceuticals, Inc., Whippany, NJ. Return to content
- Referenced with permission from the NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for Thyroid Carcinoma V.3.2024. © National Comprehensive Cancer Network, Inc. 2024. All rights reserved. Accessed July 26, 2024. To view the most recent and complete version of the guidelines, go online to NCCN.org. NCCN makes no warranties of any kind whatsoever regarding their content, use or application and disclaims any responsibility for their application or use in any way. Return to content
aPR, partial response; RAI, radioactive iodine.
References
- VITRAKVI [package insert]. Whippany, NJ: Bayer HealthCare Pharmaceuticals, Inc.; November 2023. Return to content
- Data on file. Bayer HealthCare Pharmaceuticals, Inc., Whippany, NJ. Return to content
- Drilon A, Hong DS, Van Tilburg CM, et al. Long-term efficacy and safety of larotrectinib in a pooled analysis of patients with tropomyosin receptor kinase (TRK) fusion cancer. Poster presented at: Annual Meeting of the American Society of Clinical Oncology; June 3-7, 2022; Chicago, IL. Poster 3100. Return to content
Modal - Study Design

TRIAL DESIGN FOR VITRAKVI® STUDIES
Conditional approval was based on pooled results from 3 multicenter, open-label, single-armed clinical studies1

Efficacy assessment1
192 patients with solid tumors harboring an NTRK gene fusion, ranging in age from 1 month to 84 years1,c
- All patients were required to have received prior standard therapy appropriate for their tumor type and stage of disease or who, in the opinion of the investigator, would have had to undergo radical surgery (such as limb amputation, facial resection, or paralysis-causing procedure), or were unlikely to tolerate or derive clinically meaningful benefit from available standard-of-care therapies in the advanced-disease setting1
- 92% of patients had received prior treatment for their cancer, defined as surgery, radiotherapy, or systemic therapy1
- Of these, 73% had received prior systemic therapy with a median of 1 prior systemic treatment regimen1
- 27% had received no prior systemic therapy1
- The safety population included 248 patients from Studies 1, 2, and 31
The major efficacy outcome measures were overall response rate and duration of response, as determined by a blinded independent review committee according to RECIST v1.1.1
aBrain metastases observed in 7 NSCLC, 4 thyroid, 2 melanoma, 1 SCLC, and 1 breast (nonsecretory) patient.1
bHepatocellular carcinoma.1
cAn additional 33 patients with primary CNS tumors (including astrocytoma, glioblastoma, glioma, glioneuronal tumors, neuronal and mixed neuronal-glial tumors, and primitive neuroectodermal tumor, not specified) with investigator tumor response assessment.1
CNS, central nervous system; GIST, gastrointestinal stromal tumor; NSCLC, non-small cell lung cancer; NTRK, neurotrophic tyrosine receptor kinase; RECIST, Response Evaluation Criteria in Solid Tumors; SCLC, small cell lung cancer.
References
- VITRAKVI [summary of product characteristics]. Leverkusen, Germany: Bayer AG; 2022. Return to content

Modal - Efficacy Characteristics
DEMOGRAPHIC AND PATIENT CHARACTERISTICS IN THE CLINICAL TRIAL (N=55)1,2
The clinical trials included various patient and tumor types1


aCNS, central nervous system; ECOG, Eastern Cooperative Oncology Group; GIST,
gastrointestinal stromal tumor.
References
- VITRAKVI [package insert]. Whippany, NJ: Bayer HealthCare Pharmaceuticals, Inc.; November 2023. Return to content
- Data on file. Bayer HealthCare Pharmaceuticals, Inc., Whippany, NJ. Return to content
- Drilon A, Laetsch T, Kummar S, et al. Efficacy of larotrectinib in TRK fusion–positive cancers in adults and children. N Engl J Med. 2018;378(8):731-739. Return to content
FE | Expanded Lung Orr
IN AN EXPANDED DATA SET OF NTRK GENE FUSION–POSITIVE PEDIATRIC CANCER PATIENTS (INVESTIGATOR ASSESSED, N=93)1
VITRAKVI®: HIGH RESPONSE RATES ACROSS MULTIPLE TUMOR TYPES IN PEDIATRIC PATIENTS1,2
Overall Response Rate Across Pediatric Non-CNS Tumors1

aTwo responses pending confirmation1
CNS, central nervous system; CR, complete response; NTRK, neurotrophic tyrosine receptor kinase; ORR, overall response rate; pCR, partial complete response; PR, partial response; SD, stable disease.
References
- Mascarenhas L, van Tilburg CM, Doz F, et al. Efficacy and safety of larotrectinib in pediatric patients with tropomyosin receptor kinase (TRK) fusion–positive cancer: an expanded dataset. Poster presented at: Annual Meeting of the American Society of Clinical Oncology; June 4-6, 2022; Chicago, IL. Abstract 10030. Return to content
- VITRAKVI [summary of product characteristics]. Leverkusen, Germany: Bayer AG; 2022 Return to content
FE | Expanded 80-orr
IN AN EXPANDED DATA SET OF NTRK GENE FUSION–POSITIVE LUNG CANCER PATIENTS (IRC-ASSESSED, N=26)1
VITRAKVI®: HIGH RESPONSE RATES IN NSCLC PATIENTS WITH BASELINE CNS METASTASES1


IN EVALUABLE PATIENTS WITH CNS METASTASES1
Expanded data set included 12 patients with known CNS metastases at baseline (as of July 2021).1
CNS, central nervous system; IRC, independent review committee; NSCLC, non-small cell lung cancer; NTRK, neurotrophic tyrosine receptor kinase; ORR, overall response rate; PR, partial response; SD, stable disease; TRK, tropomyosin receptor kinase.
References
- Drilon A, Lin JJ, Kummar S, et al. Updated efficacy and safety of larotrectinib in patients with tropomyosin receptor kinase (TRK) fusion lung cancer. Presented at: Annual Meeting of the American Society of Clinical Oncology; June 3-7, 2022; Chicago, IL. Abstract 9024. Return to content
FE | Expanded Differentiated Thyroid ORR
IN AN EXPANDED DATA SET OF NTRK GENE FUSION–POSITIVE THYROID CANCER PATIENTS (IRC-ASSESSED, N=30)1
VITRAKVI®: HIGH RESPONSE RATES IN NTRK GENE FUSION–POSITIVE THYROID TUMORS1
ORR in Patients With Differentiated Thyroid Cancer1

Expanded data set included 30 patients with thyroid cancer (23 differentiated; 7 anaplastic; as of July 2021)1
CR, complete response; IRC, independent review committee; NTRK, neurotrophic tyrosine receptor kinase; ORR, overall response rate; PR, partial response; SD, stable disease.
References
- Cabanillas ME, Lin JJ, Brose MS, et al. Updated efficacy and safety of larotrectinib in patients with advanced tropomyosin receptor kinase (TRK) fusion–positive thyroid carcinoma. Poster presented at: Annual Meeting of the American Thyroid Association; October 19-23, 2022; Montreal, Quebec, Canada. Abstract 108. Return to content
FE | Expanded Pediatric ORR CNS
IN AN ANALYSIS OF PEDIATRIC PATIENTS WITH NTRK GENE FUSION–POSITIVE PRIMARY CNS TUMORS (INVESTIGATOR ASSESSED, N=26)1
38% ORR IN PEDIATRIC PATIENTS WITH PRIMARY CNS TUMORS1
ORR in Pediatric Patients With Primary CNS Tumors1


DCR over ≥24 weeks was 77%
(95% CI: 56%, 91%, n=20/26) in pediatric patients1
Median OS, median PFS, and median DOR were not reached at a median follow-up of 12 months.1
aTreatment ongoing in one patient with stable disease <24 weeks.1
bTwo responses pending confirmation.1
CNS, central nervous system; CR, complete response; DCR, disease control rate; DOR, duration of response; NTRK, neurotrophic tyrosine receptor kinase; ORR, overall response rate; OS, overall survival; PFS, progression-free survival; PR, partial response; SD, stable disease.
References
- Perreault S, Doz F, Geoerger B, et al. Efficacy and safety of larotrectinib in pediatric patients with tropomyosin receptor kinase (TRK) fusion–positive primary central nervous system (CNS) tumors. Presented at: 26th Annual Meeting & Education Day of the Society of Neuro-Oncology; November 18-24, 2021; Boston, MA. Return to content
FE | Expanded Lung Orr DOR 2
IN AN EXPANDED DATA SET OF NTRK GENE FUSION–POSITIVE LUNG CANCER PATIENTS (IRC-ASSESSED, N=26)1
VITRAKVI®: HIGH RESPONSE RATES IN PATIENTS WITH NSCLC, INCLUDING THOSE WITH BASELINE CNS METASTASES1
Overall Response Rate in Patients With NSCLC1

Expanded data set included 12 patients with known CNS metastases at baseline (as of July 2021).1
CNS, central nervous system; CR, complete response; IRC, independent review committee; NSCLC, non-small cell lung cancer; NTRK, neurotrophic tyrosine receptor kinase; ORR, overall response rate; PR, partial response; SD, stable disease; TRK, tropomyosin receptor kinase.
References
- Drilon A, Lin JJ, Kummar S, et al. Updated efficacy and safety of larotrectinib in patients with tropomyosin receptor kinase (TRK) fusion lung cancer. Presented at: Annual Meeting of the American Society of Clinical Oncology; June 3-7, 2022; Chicago, IL. Abstract 9024. Return to content
